Streu-Fehler in der NIR-Spektroskopie vermeiden

Es gibt nicht nur das sichtbare Licht, also elektromagnetische Strahlung mit Wellenlängen zwischen ca. 400 nm und 780 nm, sondern noch sehr viel mehr Wellenlängenbereiche, die wir mit unseren Augen nicht mehr sehen können. Im kurzwelligen, energiereicheren Bereich folgt auf das sichtbare Licht die UV-Strahlung, und jenseits der tiefen Rottöne liegt die Infrarotstrahlung. UV-Strahlung können wir gar nicht wahrnehmen, die Infrarotstrahlung aber als Wärmeempfindung über unsere Haut fühlen. Sichtbares Licht wird in der Sensorik hauptsächlich für Messungen von Farbe und Fluoreszenz verwendet. Für die Spektrometrie von besonderem Interesse ist jedoch die Infrarotstrahlung. Sie ist deshalb so spannend, weil die meisten Moleküle auf Strahlung in diesem Wellenlängenbereich reagieren, und diese Reaktion ist messbar. Als Infrarotstrahlung (IR-Strahlung) sind Wellenlängen von 780 nm bis 1.000.000 nm (1 mm) definiert. Innerhalb dieses Bereiches gibt es noch einmal Unterteilungen. Das Nahinfrarot (NIR) ist mit Wellenlängen von 780 nm bis 2500 nm der Teil, der direkt an das sichtbare Licht anschließt.
NIR-Spektroskopie: qualitativ und quantitativ
In der NIR-Spektroskopie möchte man auf Grundlage dieser Reaktionen qualitative oder quantitative Aussagen über die beprobte Substanz treffen. Qualitativ ist eine Messung, wenn sie das Vorhandensein eines Stoffes (oder seine Abwesenheit) nachweisen kann: Worum handelt es sich bei dieser unbekannten Probe? Ist diese Substanz am Wareneingang auch die, die wir bestellt haben? Gibt es in diesem Produkt Verunreinigungen? Eine quantitative Messung trifft Aussagen über die Menge einer Substanz: Wie viel Eiweiß ist in diesen Bohnen? Wie viel Omega-3-Fettsäuren sind in diesem Pflanzenöl?
Ob pur oder Mischung, in all diesen Fällen bestehen die Proben aus bestimmten Molekülen. Wenn nun eine solche Probe mit Nahinfrarot bestrahlt wird, dann heißt das, dass Photonen mit ganz verschiedenen Wellenlängen auf die Probe treffen. Da Strahlung mit kleinen Wellenlängen energiereicher ist als Strahlung mit großen Wellenlängen, treffen hier also lauter unterschiedlich große Energieeinheiten auf die Probe. Manche von diesen Energieeinheiten entsprechend genau der Menge, die nötig ist, um ein bestimmtes Molekül zum stärkeren Schwingen zu bringen. In diesem Fall geschieht auch genau das: Die Energie des Photons wird aufgenommen ("absorbiert") und in Molekülbewegung (Schwingung der Atome gegeneinander, Verbiegen des Moleküls, Schwingung von schwach gebundenen Molekülen gegeneinander) umgesetzt. Misst man jetzt mit einem Spektrometer die Strahlung nach dem Kontakt mit der Probe, dann werden genau die Photonen fehlen, die zu Schwingungen in den Molekülen geführt haben. Die Wellenlängen dieser fehlenden, d.h. absorbierten Photonen sind die Information, mit der in der Spektrometrie gearbeitet werden kann: Jeder Stoff hat einen anderen molekularen Aufbau, und hat dadurch jeweils typische Schwingungen bei ganz bestimmten Wellenlängen. Fehlt genau diese Kombination in der Strahlung, nachdem sie Kontakt mit der Probe hatte, dann ist das ein sicheres Zeichen für das Vorhandensein dieses spezifischen Moleküls. Und aus dem Ausmaß der fehlenden Wellenlängen kann man dann auch den Anteil ablesen, den diese Substanz an der Probe hat.
Signal und Rauschen
Soweit die Theorie. In der Praxis haben wir es häufig mit komplexen Stoffgemischen zu tun. Ein Pflanzenöl besteht aus vielen verschiedenen Fettsäuren, deren chemischer Aufbau insgesamt recht ähnlich ist. Dadurch werden die jeweils typischen Absorptionskurven teilweise überlagert sein. Noch anspruchsvoller wird es bei der quantitativen Messung: Das Spektrum von Rapsöl mit 9 % Omega-3-Anteil wird mit dem einer Probe mit 10 % Anteil schwer zu unterscheiden sein. Im Feld steht deshalb häufig das Risiko im Raum, dass die kleinen Schwankungen zwischen den einzelnen Messungen vielleicht gar nicht von chemischen Unterschieden zwischen den einzelnen Proben herrühren, sondern vielleicht eher durch externe Störungen erklärt werden sollten. Ein Teil der Fehlerquellen kann durch möglichst genaue Messungen vermieden werden. Ein präzises Spektrometer ist natürlich hilfreich, aber auch ein möglichst wiederholgenauer Messaufbau ist wichtig: Zum Beispiel sollte der Messkopf konstante Abstände zu den Proben einhalten. Gleiches gilt für das Fremdlicht, also Strahlungsquellen in den Produktionsräumen, die nicht dem Zweck dienen, die Probe zu beleuchten. Im Idealfall ist die Probentemperatur über alle Messungen hinweg konstant, auch die Oberfläche und Struktur des untersuchten Materials immer möglichst identisch, die Umgebungstemperatur variiert nicht usw. usf. Außerdem ist vorab die möglichst präzise chemische Analyse der Proben wichtig. Meist wird im Labor mit klassischen chemischen Methoden der Gehalt der interessanten Stoffe in der Probe analysiert (Bei gebräuchlichen Stoffen liegt dieser Schritt jedoch schon Jahrzehnte in der Vergangenheit, und stattdessen wird ein präzises, bereits kalibriertes Laborspektrometer eingesetzt). Die Ergebnisse dieser Analysen (die Referenzdaten) werden dann anschließend mit spektralen Messungen abgeglichen, so dass mathematisch herausgearbeitet werden kann, welche Kurvenverläufe auf welche Inhaltsstoffe zurückzuführen sind. Zukünftig reicht dann die spektrale Information, um den Gehalt des Inhaltsstoffs bestimmen zu können.
Aber selbst, wenn all diese Punkte geflissentlich bearbeitet wurden, wird es immer noch optische Effekte geben, die die chemischen Informationen im Spektrum überlagern, und teilweise sogar den Großteil der Unterschiede zwischen einzelnen Messungen erklären. Bei diesen optischen Effekten handelt es sich fast immer um Streuung.
Reflexion und Brechung
Streng genommen findet immer Streuung statt, wenn elektromagnetische Wellen auf Materie treffen: Photonen mit passender Energie werden in diesem elastische Streuung genannten Fall absorbiert und gleichzeitig, mit identischer Wellenlänge wieder reemittiert (das ist ein anderer Vorgang als derjenige, bei dem die Moleküle entlang ihrer Atomverbindungen zum Schwingen gebracht werden). Auf Makroebene können mit der elastischen Streuung auch Phänomene wie Reflexion und Brechung erklärt werden, die aus der klassischen geometrischen Optik bekannt sind. Diese Form der Streuung ist grundsätzlich für die Spektrometrie auch nicht unwichtig, weil sie zu diffuser Reflexion führt.
Gerichtete und diffuse Reflexion
In der geometrischen Optik wird von einer idealisierten Situation ausgegangen: Es gibt ein festes Material mit glatter Oberfläche, so dass eingehende Lichtstrahlen in genau dem Winkel reflektiert werden, mit dem sie auch auf die Oberfläche trafen. Das ist gerichtete Reflexion. Zoomen wir jetzt auf Atomebene an den Ort des Geschehens, dann zeigt sich, dass die vermeintlich feste, glatte Oberfläche aus lauter mehr oder weniger eng und systematisch angeordneten Molekülen besteht, zwischen denen nichts ist. Es ist also nicht garantiert, dass jedes Photon den Weg der gerichteten Reflexion einschlägt. Stattdessen kann es auch nach innen in die Probe gestreut werden, wieder auf ein Molekül treffen, wieder gestreut werden und erst nach einem längeren Weg durch das Material wieder nach außen gelangen. Dieser Fall wird diffuse Reflexion genannt. Bei der gerichteten Reflexion findet praktisch keine Interaktion zwischen Material und Strahlung statt. In diesem Fall kann die im Anschluss gemessene Strahlung auch fast nichts an chemischer Information enthalten. Gerichtete Reflexion kann also den Teil der Strahlung, der die gesuchten Absorptionssignale enthält, überlagern, und wird deshalb in der Spektrometrie als Störfaktor gedeutet und möglichst vermieden. Messtechnisch interessant ist die diffuse Reflexion, denn hier ist die Strahlung vor der Messung mit dem Spektrometer in die Probe eingedrungen. Dabei wird es zu Absorption kommen, so dass die reflektierte Strahlung Informationen über den Aufbau des Materials enthält.
unerwünschte Streueffekte in der Spektrometrie
Die Streueffekte, die zu diffuser Reflexion führen, bilden eine wesentliche Grundlage für die NIR-Spektroskopie. Andere Streueffekte sind hauptsächlich Quellen für störendes Rauschen, das die interessante chemische Information potenziell verdecken.
Rayleigh-Streuung
Rayleigh-Streuung tritt auf, wenn die Partikel deutlich kleiner als die Wellenlängen der Strahlung sind (ca. 1/10 der Wellenlänge). Das trifft zum Beispiel auf das sichtbare Sonnenlicht und die Atome und Moleküle in unserer Atmosphäre zu. Diese Teilchen haben keine Resonanzfrequenzen im Wellenlängenbereich des sichtbaren Lichts. Wenn sie von einem Photon angeregt werden, dessen Wellenlänge deutlich größer als sie selbst ist, dann ist das ein Fall elastischer Streuung: Sie nehmen das Photon auf, oszillieren dadurch stärker, hören gleichzeitig wieder auf und reemittieren das Photon mit identischer Frequenz/Wellenlänge. Die Atome sind in de Atmosphäre zufällig ausgerichtet, und deshalb werden sie das Licht grundsätzlich auch zufällig streuen. Das Ausmaß der Richtungsänderung ist aber auch von der Wellenlänge des Photons abhängig: Je näher es an der Resonanzfrequenz liegt, die jedes Atom im UV-Bereich hat, desto stärker wird das reemittierte Photon von der ursprünglichen Richtung abweichen. Genau das passiert an unserem Himmel: Das kurzwellige, blaue Licht wird viel wahrscheinlicher von der ursprünglichen Richtung abweichen, auf neue Atome treffen, wieder gestreut werden - bis es uns als irdischen Beobachter:innen so erscheint, als würde der gesamte Himmel blau sein, weil von jeder Himmelsrichtung das gestreute blaue Licht auf unsere Netzhaut trifft. Rayleigh-Strahlung kann auch mit größeren Objekten stattfinden, solange dann auch die Wellenlängen im gleichen Verhältnis wachsen. Rayleigh-Streuung kann also grundsätzlich auch an winzigen Fasern, Tröpfchen oder Blasen stattfinden, solange die Wellenlänge der einfallenden Strahlung Durchmesser von mind. dem zehnfachen dieser Teilchen beträgt. Die Form der Teilchen ist dabei wenig von Belang.
Mie-Streuung
Je mehr sich die Partikelgröße der Wellenlänge annähert, desto weniger lassen sich die beobachtbaren Phänomene mit Rayleigh-Streuung erklären. Sobald die Wellenlängen ungefähr dem Partikeldurchmesser entsprechen und die Partikel ungefähr kugelförmig sind, sind wir im Terrain der Mie-Streuung angelangt. In diesem Bereich ist die Streuung weit weniger wellenlängenabhängig. Auch das kann man wieder am Himmel sehen, denn Mie-Streuung ist der Grund, warum Wolken so grau wirken. Die winzigen Wassertröpfchen sind ungefähr rund und entsprechen in ihrer Größe ungefähr der Wellenlänge des sichtbaren Lichts. Dadurch werden die Lichtstrahlen der Sonne an ihnen wellenlängenunabhängig gebrochen. Alle ursprünglich im Sonnenlicht enthaltenen Farbanteile bleiben also erhalten, sie werden jedoch zum Teil weg von unserem Auge gestreut, so dass diese Stelle des Himmels dunkler erscheint. Werden die Partikel deutlich größer als die Wellenlängen, wieder ungefähr um den Faktor 10, dann verlassen wir den Bereich der Mie-Streuung. Ab diesem Moment können die Effekte mit den Mitteln der geometrischen Optik gut vorhergesagt werden. Im Nahinfrarotbereich ist es durchaus nicht unwahrscheinlich, bei spektrometrischen Messungen direkt mit Mie-Streuung konfrontiert zu sein. Kleinste Fasern, Fetttröpfchen, Luftbläschen oder ähnliche Partikel mit Durchmessern von 1-3 µm können sich durchaus in den Proben befinden, die mit NIR-Spektrometern untersucht werden.
Das ist für präzise Ergebnisse problematisch, denn diese Streueffekte werden zu veränderten Spektren führen: Nun haben wir nicht mehr nur Absorption als Grund, warum auf bestimmten Wellenlängen weniger Strahlung gemessen wird, sondern auch Streueffekte.
Spektrale Vorverarbeitung
Diese physikalische Verschleierung des chemischen Signals waren historisch tatsächlich eine Einschränkung, die den produktiven Einsatz der NIR-Spektroskopie zunächst verhindert haben. Erst die mathematische Aufbereitung der Spektren mit Hilfe von Computern brachte hier Fortschritt.
Ableitungen
Die erste und noch immer genutzte Form der Vorverarbeitung war die Ableitung. Ableitungen betonen die Peaks in Spektren. Höhere Ableitungen können ggf. auch Peaks auftrennen, die durch Überlagerung der Einzelpeaks verschiedener Substanzen entstanden sind. Bei der ersten Ableitung wird aus einem Peak eines Absorptionsspektrums ein Aufschwingen zu einem Maximum kurz vor und ein entsprechend gespiegeltes Minimum kurz hinter dem ursprünglichen Peak, so dass die Stelle des ursprünglichen Peaks exakt 0 beträgt. Die zweite Ableitung führt zu einem Minimum an der Stelle, an der im unbearbeiteten Spektrum ein Maximum lag. Diese Ableitung wird in der Praxis wohl am häufigsten genutzt.
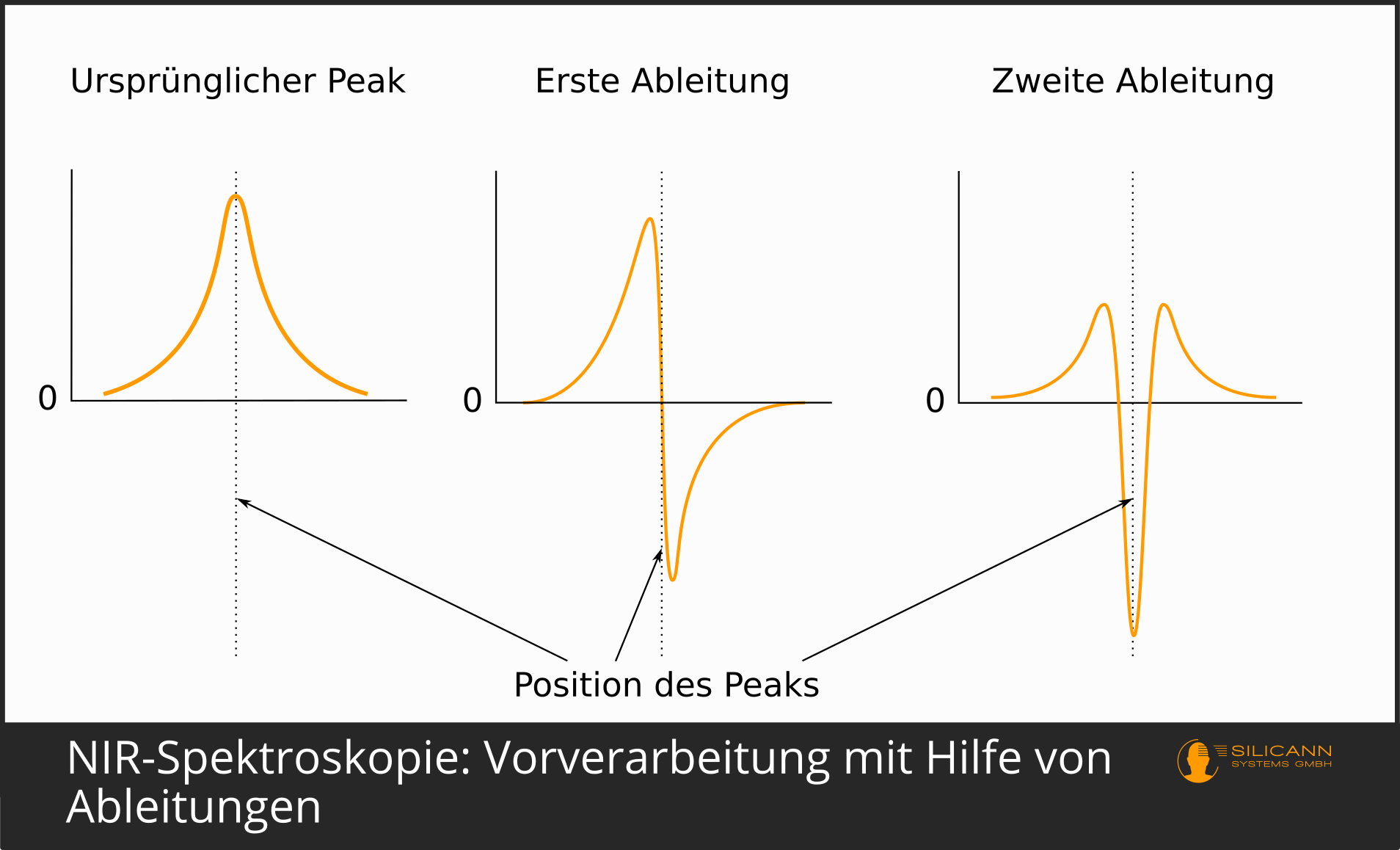
Auch die Nutzung der dritten und vierten Ableitung ist nicht unüblich. Es gilt jedoch, dass mit der Nutzung höherer Ableitungen auch eher Artefakte eingeführt werden, die künstlich Peaks erfinden, die im tatsächlichen Spektrum gar nicht vorhanden waren. Ein sehr häufig eingesetzter Algorithmus ist Savitzky-Golay.
Ableitungen arbeiten mit einem einzelnen Spektrum, ohne Bezug auf die anderen Spektren der Messreihe.
MSC: Multiplicative Scatter Correction
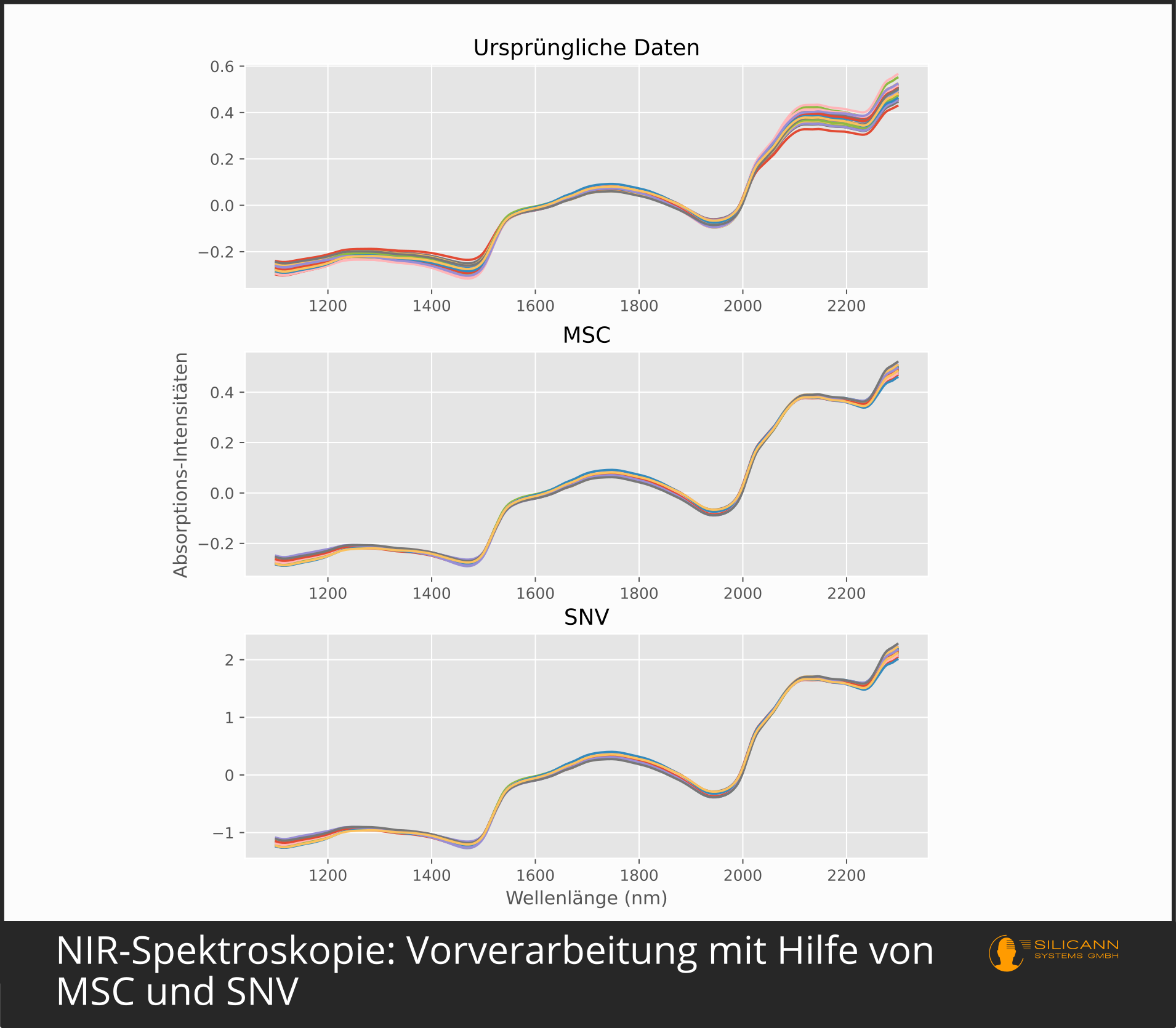
Das ist bei der Multiplicative Scatter Correction anders. MSC soll sowohl bei additiven als auch bei multiplikativen Fehlern das Signal vom Rauschen trennen. Additive Fehler sind konstante Streufehler. Sie würden bei einem Spektrum dazu führen, dass sämtliche Werte um einen bestimmten Faktor nach oben oder unten entlang der y-Achse, der Achse mit den Intensitäten, verschoben sind. Eine mögliche Ursache dafür wären etwa durch Streuung verursachte Pfadlängenunterschiede. Multiplikative Fehler führen zu einer Neigung des Spektrums, so dass die Werte immer größer werden, je weiter wir in den langwelligen Bereich der x-Achse, der Achse mit den Wellenlängen, kommen. Diese Art von Fehler wird z.B. durch Mie-Streuung an kleinen Partikeln verursacht, die sich in dieser Größenordnung befinden. Basis der Funktionsweise der Multiplicative Scatter Correction ist die Beobachtung, dass die Streueffekte, verursacht z.B. diese Mie-Streuung, eine andere Wellenlängenabhängigkeit aufweisen als die Absorptionseffekte, die sich durch die chemische Beschaffenheit der Probe ergeben. Das konkret gemessene Spektrum soll dazu mit dem idealen Spektrum der Probe, einem Spektrum ohne Streufehler welcher Art auch immer, verglichen werden. So ein ideales Spektrum ist in der Praxis schwer zu beschaffen, darum wird in der Praxis fast immer der Weg aus dem Original-Paper eingeschlagen: Aus vielen verschiedenen Messungen sehr ähnlicher Proben wird ein gemitteltes Spektrum berechnet und als Ersatz für das ideale Spektrum genutzt. Intern werden dann per Least-Squares-Regression a und b für folgende Formel berechnet:
X = a + b*X' + E
Dabei ist X das konkrete Spektrum, X' das ideale Spektrum, a die additive Komponente (d.h. der additive Fehler, die Verschiebung entlang der y-Achse) und b die multiplikative Komponente (die Steigung um den Schnittpunkt mit der y-Achse). E entspricht dem Rest-Spektrum, das dann im Idealfall vollständig chemische Informationen enthält. Schnittpunkt und Steigung werden dabei so berechnet, dass das konkrete Spektrum und das gemittelte Spektrum mit derselben Absorption bei der kürzesten Wellenlänge starten und im weiteren Wellenlängenverlauf dieselbe Steigung aufweisen. Seit der Publikation des einführenden Papers im Jahre 1985 wird MSC sehr häufig als Vorverarbeitungsschritt eingesetzt, besonders in Kombination mit PLS als Kalibrationsmethode. Mittlerweile gibt es auch verschiedene Erweiterungen dieses Ansatzes, die dann insb. auch komplexere, nichtlineare Streuungen kompensieren können.
SNV: Standard Normal Variate Transformation
Die Vorverarbeitungsvariante Standard Normal Variate (SNV) wird für ähnliche Situationen eingesetzt wie MSC: als Mittel gegen Streuung und für Szenarien mit variierender Pfadlänge zur Probe. Entsprechend kann es auch zur Kompensation unterschiedlich rauer Probenoberflächen genutzt werden, denn das ist ja gleichbedeutend mit unterschiedlichen Partikelgrößen, und damit wieder (Mie-)Streueffekten. Im Unterschied zu MSC orientiert sich die Korrektur bei SNV jedoch nur am einzelnen, konkreten Spektrum. Die Autor:innen des Papers begründen das damit, dass die multiplikativen Streueffekte ihre Ursache in der jeweils individuellen Probe haben (z.B. in der individuell verschiedenen Oberflächenbeschaffenheit), nicht in der Gesamtheit aller Proben, und deshalb bei einer Korrektur auch nur das einzelne Spektrum herangezogen werden muss. SNV reduziert den Fehleranteil der spektralen Intensitäten, indem der Mittelwert aller Intensitäten gebildet, von allen Einzelwerten abgezogen und anschließend durch die Standardabweichung dividiert wird. Nach Anwendung von SNV hat das resultierende Spektrum in der Regel noch immer einen klar erkennbaren steigenden Trend in Richtung größerer Wellenlängen. Die Autor:innen haben dafür ein Detrending mit Hilfe eines Polynoms zweiten Grades vorgeschlagen.
MSC oder SNV?
In der Praxis liefern MSC und SNV in der NIR-Spektroskopie im Allgemeinen vergleichbare Ergebnisse. Welchem Ansatz der Vorzug zu geben ist, und ob ggf. auch schon die zweite Ableitung reicht, hängt stark von den konkreten Proben und den Messbedingungen ab. Da Ableitungen primär die Peaks betonen und MSC, SNV und ähnliche Algorithmen primär Fehler reduzieren, spricht auch grundsätzlich erst einmal nichts gegen die gleichzeitige Anwendung beider Ansätze.
Der Vorverarbeitung des Spektrums folgt ja meist die Kalibration, d.h. der Versuch, aus dem optischen Signal die chemische Information vorhersagen zu können. Im Zweifel sollte sich die Entscheidung der Vorverarbeitungsmethode am resultierenden Vorhersagefehler dieses Kalibration orientieren.
Quellen
- Hecht: Optics, 5th edition, 2017
- Geladi, MacDougall, Martens: Linearization and Scatter-Correction for Near-Infrared Reflectance Spectra of Meat, 1985
- Barnes, Dhanoa, Lister: Standard Normal Variate Transformation and De-trending of Near-Infrared Diffuse Reflectance Spectra, 1989


